





The Mission Bio Tapestri platform enables the high-throughput analysis of DNA and protein targets simultaneously across thousands of individual cells, but it can do far more than that. With simple modifications, we can use Tapestri to elucidate the complex relationship between genetic alterations and cellular phenotypes in patient samples. In this blog, we’re shining a spotlight on some of Mission Bio customers who have adapted the Tapestri instrument for multi-omics workflows, including the addition of targeted single-cell RNA sequencing and even DNA methylation profiling.
The Tapestri Platform: Beyond Single-Cell DNA Sequencing
The original Tapestri microfluidic platform was designed for single-cell DNA sequencing (scDNA-Seq) and surface protein analysis, connecting genotype to immunophenotype. However, there are several layers of important information or ‘color’ between the genetic blueprint and the proteins expressed by cells. This includes the transcriptome, which is the collection of RNA transcripts within a cell. Before we discuss customer adaptations to Tapestri, let’s take a look at single-cell RNA sequencing, how it can provide vital information about gene expression, and the current challenges of combining single-cell RNA sequencing and DNA sequencing workflows.
What is single-cell RNA-Seq?
Single-cell RNA sequencing, or scRNA-Seq, is a method for the detection and quantitative analysis of RNA transcripts within single cells.1 This allows researchers to profile the expression of genes, providing a necessary link between genotype and phenotype in individual cells. Single-cell RNA sequencing of the transcriptome can be used to study gene expression in cell therapy products, heterogeneous tumor samples, blood stem cells, and other tissues.2
Why use both scRNA-Seq and scDNA-Seq in your research?
Multi-omics, which combines the sequencing of multiple ‘omes—like the genome and transcriptome—can help elucidate the relationships between these different layers of biological information.3 In biomedical research, single-cell multi-omics, including single-cell RNA sequencing and DNA sequencing, allows researchers to develop a more complete understanding of cell biology and the effects of natural genetic variation or genetic engineering on gene expression and cellular phenotypes.
Simultaneous sequencing of the genome and transcriptome from thousands of single cells has a wide range of exciting biological applications. For example, it can be used to study the complex tumor microenvironment, the process of hematopoiesis, the effects of genetic mosaicism, clonal fitness, and the biological process of aging. It is also particularly relevant in the development of safe and effective cell and gene therapies, which are inherently variable and often contain multiple genetic edits.4 By integrating targeted scRNA-seq and scDNA-Seq, we can deepen our understanding of human development, health, aging, and disease.
Challenges in integrating scDNA-Seq and scRNA-Seq workflows
Integrating single-cell RNA sequencing into a multi-omics workflow might seem like a no-brainer, but it’s not without challenges. Currently, methods for the simultaneous sequencing of both genomic DNA and RNA are low-throughput and labor-intensive, while high-throughput technologies are less sensitive and cannot provide independent readouts of these targets. Ideally, single-cell RNA sequencing and single-cell DNA sequencing would be performed on a sensitive, high-throughput platform, but this presents a key obstacle.
To understand why, let’s explore how Mission Bio’s Tapestri instrument works. The Tapestri instrument is designed for single-cell DNA sequencing and protein analysis. It works by enclosing individual cells in a droplet containing lysis buffer, which breaks down the cell to expose the nucleic acids, and proteinase K, which removes protein contaminants. After heat-inactivation of the enzyme, these cell-filled droplets are merged with droplets containing PCR reagents and barcodes, allowing the target DNA to be amplified while attached to an identifier. This enables researchers to identify either natural genomic variants or introduced edits across a pool of cells in a high-throughput fashion.
However, the enzymatic reactions required for DNA sequencing are not compatible with single-cell RNA sequencing methods, making it difficult to sequence both the DNA and RNA from the same individual cells. Fortunately, some clever solutions have allowed for both targeted scDNA-Seq and scRNA-Seq to be performed on our Tapestri platform. Let’s explore some of the different studies that have merged these two technologies using the Tapestri device.
scRNA-Seq and scDNA-Seq Allow Functional Phenotyping of Genomic Variants
In a recent study published in Nature Methods, a team of researchers from the European Molecular Biology Laboratory (EMBL) and Stanford University School of Medicine aimed to profile the functional effects of both coding and noncoding variants on gene expression.5 Using the Tapestri platform, the team developed single-cell DNA-RNA sequencing (SDR-Seq), a robust method that simultaneously performs single-cell RNA sequencing and DNA sequencing. This is made possible by performing in situ reverse transcription in the cells to create cDNA before the typical Tapestri workflow begins.
Proof-of-principle in induced pluripotent stem cells
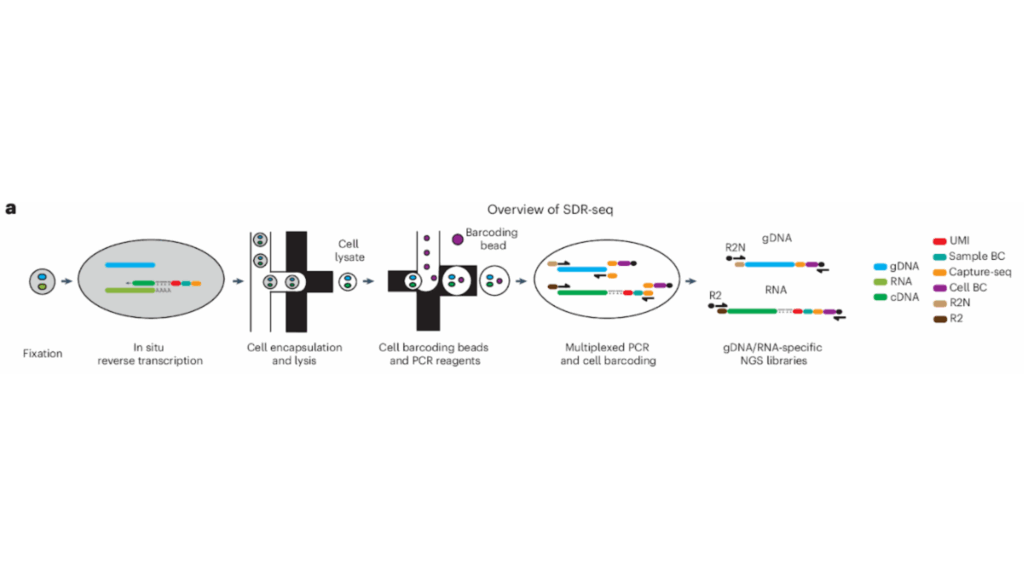
An overview of the SDR-Seq workflow, which integrates single-cell RNA sequencing into the Tapestri platform; in situ reverse transcription occurs before loading onto the device. The SDR-Seq method enables the study of coding and noncoding variants and their effects on gene expression. Adapted from: Lindenhofer D, et al. Nat Methods (2025).5
As proof-of-principle, the authors demonstrated the SDR-Seq workflow on human induced pluripotent stem (iPS) cells, with 28 targets from DNA and 30 from RNA. Cultured cells were dissociated into a suspension, then fixed. Using custom poly(dT) primers, the team performed in situ reverse transcription, adding a unique molecular identifier (UMI), a barcode (BC), and a capture sequence (CS) to each cDNA molecule.
At this stage, the cells contained both cDNA and genomic DNA (gDNA). They were loaded onto the Tapestri instrument, where they were mixed with droplets containing lysis buffer, proteinase K, and reverse primers for each target. These droplets of cells are then added to droplets containing PCR reagents, forward primers with CS overhangs, and barcoding beads. Barcoding beads contained unique BC oligos with overhangs that matched the CS overhangs of the forward primers.
Using multiplexed PCR, the authors then amplified the gDNA and cDNA targets from the cells, barcoding them for further analysis and generating distinct libraries that were separated for sequencing. Sequencing analysis showed that this high-throughput approach was able to detect DNA and RNA targets with high sensitivity across thousands of cells. The authors next tested SDR-Seq on expanded panels of hundreds of gDNA and RNA targets, showing that this method is highly sensitive and reproducible even at much larger scales.
The effects of coding and noncoding variants
Further experiments demonstrated that SDR-Seq is a robust method for detecting changes in gene expression mediated by CRISPR inhibition (CRISPRi). This method could also confidently detect even subtle changes in gene expression mediated by the insertion of expression quantitative trait loci (eQTL) variants via prime editing. Finally, the authors used base editing to introduce eQTLs, including noncoding variants, revealing that several of these variants had a significant effect on the expression of the target genes.
These comprehensive analyses show that by integrating single-cell RNA sequencing, the Tapestri platform can be used to detect both coding and non-coding genetic variants in single cells and associate them with changes in gene expression with a high level of confidence.
Testing in primary B cell lymphoma samples
Identifying causal links between genetic variants and gene expression is vital in cancer research, providing a deeper understanding of tumorigenesis and cancer progression. As such, the authors wanted to put their SDR-Seq method to the test in cryopreserved primary B cell lymphoma samples, analyzing between 2,600 and 8,400 cells per patient. Combining single-cell RNA sequencing and DNA sequencing on the Tapestri platform allowed the team to profile the effects of genetic variants on gene expression and differentiation within tumors.
The SDR-Seq method was able to identify that cells with a higher mutational burden displayed enhanced tumorigenic gene expression and elevated B cell receptor signaling. This study clearly demonstrates that combining single-cell RNA sequencing and DNA sequencing on the Tapestri platform has strong potential in the study of the complex tumor microenvironment and the evolution of cancer. Watch coauthor Julia Bauman explain the paper in 60 seconds here.
Using scRNA-Seq and scDNA-Seq to Understand Clonal Mosaicism
Another key application of combined single-cell RNA sequencing and DNA sequencing on the Tapestri platform is in the study of clonal mosaicism. Somatic or clonal mosaicism refers to the genetically distinct populations of cells within an individual; cells gain post-zygotic genetic mutations and can accumulate throughout a person’s life via clonal expansion. This phenomenon is common in the biological process of aging, but it is also a hallmark of cancer.
Identifying the links between the genetic driver mutations and the phenotype of these cells can help us to understand how clonal mosaicism affects human health, chronic disease, and cancer. Bulk sequencing of tissue samples can determine how often specific mutations occur, but fails to elucidate clonal architecture and how driver mutations affect cellular phenotypes.
A recent preprint published on BioRxiv by researchers from Weill Cornell Medicine used the Tapestri platform to develop single-cell Genotype-to-Phenotype sequencing (scG2P).6 scG2P is a high-throughput, multiplexed method for the joint capture of recurrent DNA mutations and mRNA transcripts in single cells isolated from solid tissue. In this case, the samples analyzed were phenotypically normal esophageal (PNE) biopsies from individuals with high exposure to tobacco and alcohol.
Co-capture of somatic mutations and mRNA transcripts
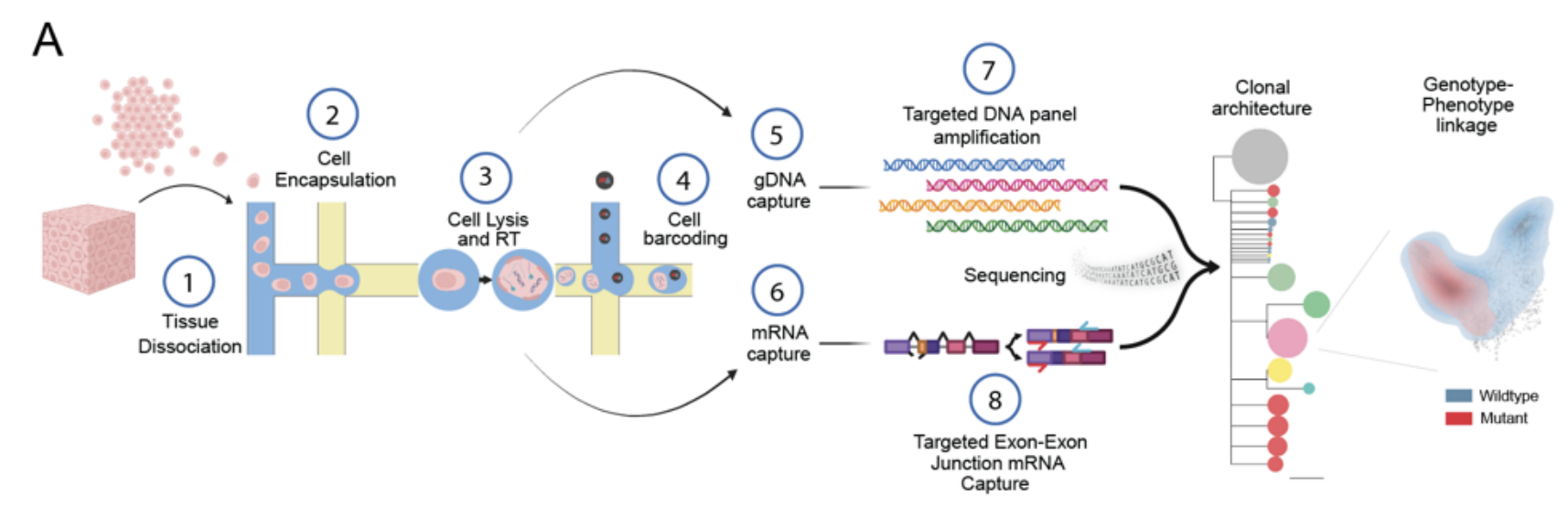
A schematic of the scG2P method, which performs reverse transcription of RNA after formation of the first droplet in the Tapestri workflow to allow for single-cell RNA sequencing. This approach allows researchers to study clonal architecture and link genotype to phenotype.
Adapted from: Yuan DJ, et al. bioRxiv (2024).6
Similar to the SDR-Seq paper we discussed above, this study also employs reverse transcription of mRNA targets to perform single-cell RNA sequencing, however, the methods also differ in several ways. In the scG2P approach, reverse transcription of mRNA targets occurs during the formation of the first droplet, after dissociated cells are loaded onto the Tapestri platform. By adding capture handles to the cDNA amplicons, the authors allow for downstream barcoding.
During formation of the second droplet, cell barcodes are added to gDNA and cDNA targets, followed by multiplexed PCR for amplification. The barcoded DNA and RNA amplicons were separated by adding an RNA biotin oligo to the final PCR product and then using magnetic streptavidin-coated beads; the biotin oligo forms a complex with RNA targets and then binds strongly to the streptavidin, allowing the RNA amplicons to be magnetically separated from the rest of the product.
Importantly, the RNA capture primers were designed to cover exon-exon junctions, allowing them to be easily differentiated from off-target capture of gDNA. While other studies use scDNA-Seq panels that focus on specific hotspots where mutations are known to occur, this paper targeted amplicons across entire genes to enable a deeper understanding of the mutational landscape.
The authors explored 118 genomic amplicons across six known driver genes, including NOTCH1, TP53, NOTCH2, NOTCH3, FAT1, and PPM1D, as well as 86 RNA targets that serve as phenotypic markers. In total, more than 5000 cell nuclei were isolated and analyzed across PNE samples from five donors of different ages.
Studying clonal mosaicism in the esophageal epithelium
After validating SDR-Seq in mixed esophageal and colon cells, the authors applied the approach to nuclei isolated from the cryopreserved PNE samples. The results showed that between 31 and 89 percent of cells had at least one mutation. However, it was exceedingly rare for cells to have more than one mutation, indicating that the majority of cell clones in the PNE were driven by a single mutation.
Mutations in NOTCH1 and TP53 dominated the clonal landscape in PNE, being associated with larger clonal expansions than mutations in the other target genes. The authors also noted that the increasing age of samples was associated with an increase in clonal diversity, suggesting that different clones compete with each other in aging PNE tissue.
Genotype-to-phenotype mapping
After studying the gDNA mutations, the team wanted to perform genotype-to-phenotype mapping, investigating the mechanisms that endow some clonal populations with a selective advantage. This is where single-cell RNA sequencing comes into play. Cells were sorted based on canonical marker genes and were given cycling and differentiation scores based on their expression of RNA markers, allowing the authors to place each cell on a continuum of cell cycle and differentiation.
This approach showed that clones with TP53, NOTCH1, and NOTCH3 mutations were biased towards early states of differentiation. While TP53 mutants also showed increased cell cycling, NOTCH1 clones were only biased towards early differentiation. In contrast, the rare double-mutant clones were biased towards increased proliferation. These results indicate that NOTCH1 driver mutations—which are less common in esophageal cancer—may have a protective role against tumorigenesis. However, the strong phenotypic differences and increased proliferation of the rare double-mutant clones support the hypothesis that multiple driver mutations lead to malignant transformation.
Together, these results of this study demonstrate that combining single-cell RNA sequencing and DNA sequencing on the Tapestri instrument enables the reconstruction of clonal hierarchies in solid tissues and can jointly capture genotype and phenotype with high accuracy, providing valuable insights into clonal mosaicism and its effects on health and disease.
Tracking Clonal Differentiation Trajectories in Haematopoiesis via DNA Methylation and scRNA-Seq
As humans age, we experience clonal expansions in our stem cells, with some clones dominating the production of blood and others vanishing. By tracking stem cell clones throughout the journey of hematopoiesis we can gain valuable insights into human development, aging, and disease.
However, there are several key challenges that limit the widespread application of lineage tracing. In animal models, for example, lineage tracing is achieved via complex genetic engineering, which cannot be used in human subjects or to study the natural process of aging. In contrast, lineage tracing in human samples relies on endogenous clonal markers like somatic mutations, which are sparse and may not accurately reflect clonal identity. Additionally, these methods have limited throughput.
To overcome these obstacles, researchers from the Barcelona Institute of Science and Technology developed a high-throughput method that uses DNA methylation and single-cell RNA sequencing analysis to track and characterize stem cell clones in blood samples. In a recent study published in Nature, the authors describe adapting the Tapestri platform for DNA methylation analysis and combining this with the SDR-Seq method we discussed above.7
Mapping hematopoiesis via DNA methylation
The authors aimed to track clonal identity and cell state using DNA methylation data, which has previously been associated with clonal signals in bulk analyses of malignant and healthy tissues. To do so, the authors incorporated a DNA methylation-sensitive restriction enzyme (HhaI) into the Tapestri workflow to digest non-methylated DNA. This allowed them to retain and sequence only methylated CpG sites (epi-mutations) for the amplification step. Named scTAM-Seq (single-cell targeted analysis of the methylome), the authors applied this approach in both mouse and human samples, examining hundreds of DNA methylation amplicons.
After validating the method in lentiviral-barcoded mouse hematopoietic stem cells, the team was able to create a methylation-based map of mouse stem cell differentiation. The results showed that DNA methylation encodes both clonal identity and cell state, and surface protein expression data confirmed that some CpG sites are associated with differentiation. Using their EPI-Clone algorithm, the authors analyzed clonal identity in mouse stem cells, mature immune cells, and other tissues such as the lung, demonstrating the broader applications of DNA methylation as a clonal barcode.
Combining DNA methylation analysis with single-cell RNA sequencing
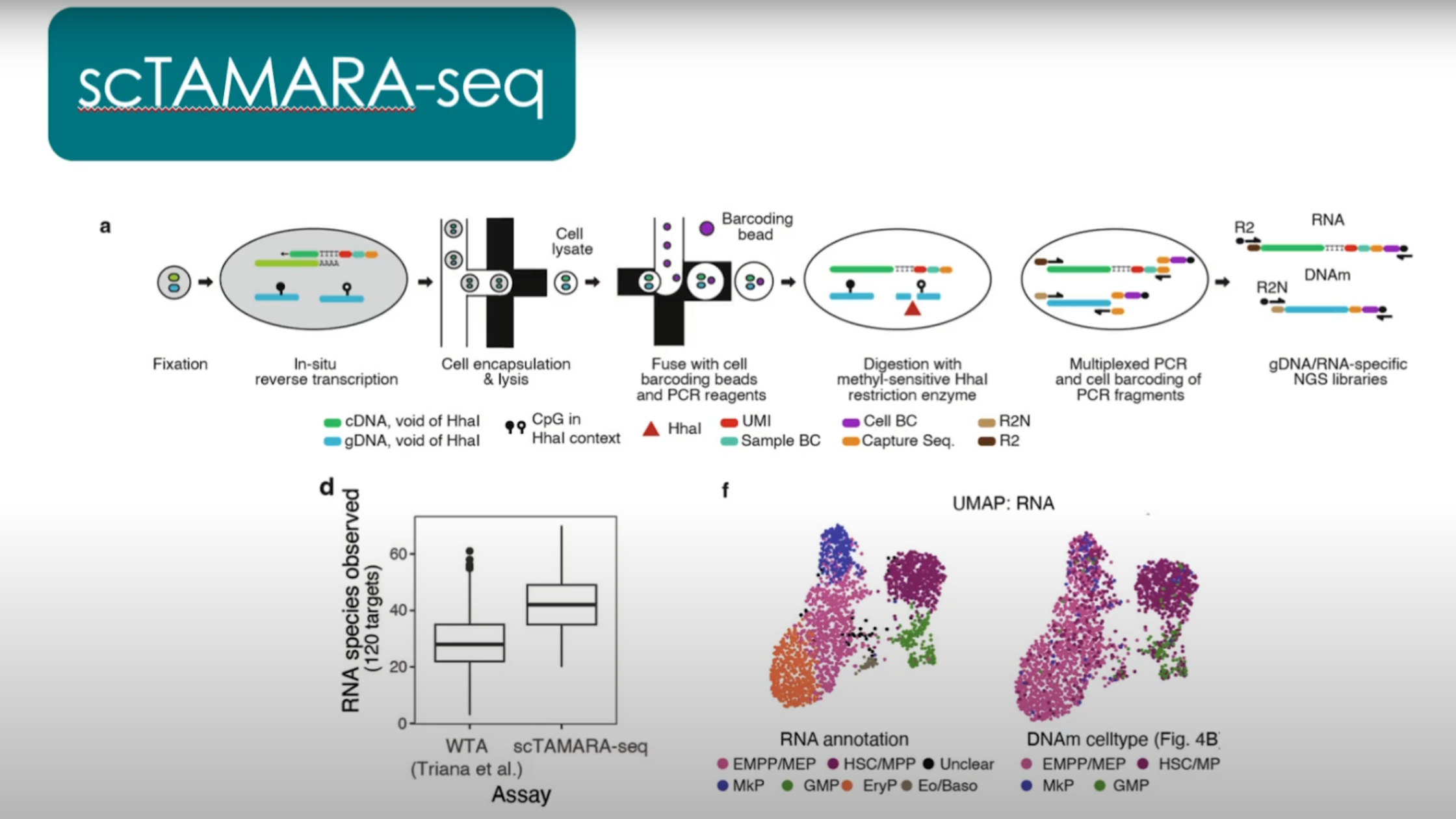
Caption: The scTAMARA-seq method incorporates both single-cell RNA sequencing and DNA methylation analysis on the Tapestri device, allowing researchers to map the process of hematopoiesis and track clonal identity and cell state.
Adapted from: Velten L. Tracing blood stem cell clones with somatic epimutations (2025) https://www.youtube.com/watch?v=KNsCGkNDU0s
Finally, the authors wanted to resolve transcriptional differences between distinct clones identified in human bone marrow samples. To do so, they combined their scTAM-Seq approach with the SDR-Seq method described by the team at Stanford and EMBL, creating scTAMARA-Seq (single-cell targeted analysis of the methylome and RNA). Together, these methods encompass single-cell RNA sequencing, DNA sequencing, methylation data, and surface protein expression.
With 120 RNA targets, the team used single-cell RNA sequencing analysis to confirm that cell-state annotations based on DNA methylation data were accurate. They also revealed that clonal differentiation is correlated with changes in gene expression in early stem and progenitor cell states. To learn more about the study, you can watch a presentation by lead author Lars Velten on the Mission Bio YouTube channel here.
Applying Single-Cell RNA Sequencing and DNA Sequencing in Your Own Research
Although originally designed for scDNA-Seq and protein expression analysis, the strength of Mission Bio’s Tapestri platform is its adaptability. The studies we’ve discussed in this article showcase the different ways Tapestri can be modified and adjusted for expanded multi-omics workflows, including single-cell RNA sequencing analysis and even DNA methylation data.
These innovative strategies have allowed our customers to fill the critical gaps between genotype and phenotype, elucidating the effects of coding and noncoding variants of gene expression, studying clonal mosaicism in solid tissues, and even tracing lineages of stem cells and their differentiation states throughout hematopoiesis.
By applying targeted single-cell RNA sequencing and DNA sequencing at the single-cell level rather than in bulk samples, no critical information is lost, and the high-throughput, cost-efficient nature of the Tapestri platform allows this with ease. With predesigned panels of DNA, RNA, and protein markers available, or build custom, fit-for-purpose assays through our Pharma Assay Development (PAD) services, no target is off-limits. If you’d like to generate deeper disease insights by integrating single-cell RNA sequencing, DNA sequencing, and more into your own research, let’s talk.
References
- Haque A, Engel J, Teichmann SA, Lönnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017;9(1):75. doi:10.1186/s13073-017-0467-4
- Jovic D, Liang X, Zeng H, Lin L, Xu F, Luo Y. Single‐cell RNA sequencing technologies and applications: A brief overview. Clin Transl Med. 2022;12(3):e694. doi:10.1002/ctm2.694
- Chen C, Wang J, Pan D, et al. Applications of multi‐omics analysis in human diseases. MedComm. 2023;4(4):e315. doi:10.1002/mco2.315
- Goss K, Horwitz EM. Single-cell multiomics to advance cell therapy. Cytotherapy. 2025;27(2):137-145. doi:10.1016/j.jcyt.2024.10.009
- Lindenhofer D, Bauman JR, Hawkins JA, et al. Functional phenotyping of genomic variants using joint multiomic single-cell DNA–RNA sequencing. Nat Methods. Published online September 1, 2025:1-10. doi:10.1038/s41592-025-02805-0
- Yuan DJ, Zinno J, Botella T, et al. Genotype-to-phenotype mapping of somatic clonal mosaicism via single-cell co-capture of DNA mutations and mRNA transcripts. bioRxiv. Preprint posted online May 23, 2024:2024.05.22.595241. doi:10.1101/2024.05.22.595241
- Scherer M, Singh I, Braun MM, et al. Clonal tracing with somatic epimutations reveals dynamics of blood ageing. Nature. 2025;643(8071):478-487. doi:10.1038/s41586-025-09041-8

