





Modeling Cancer is Challenging
Cell- and animal-based models are integral to progress in understanding and treating genetic diseases. By generating the underlying mutations in cells, organoids, or whole organisms, researchers can effectively recreate genotypic and phenotypic characteristics of the disease of interest. Such models are key for understanding pathogenesis and for testing the efficacy and safety of new therapies. For many genetic diseases, modeling is typically accomplished by making genetic changes to cells and then analyzing them at the population, or bulk, level. With such an approach, characteristics are measured in an entire cell sample and represent an “average” of the cell population.
Yet, we know that for some diseases — such as cancer — the cell-to-cell variability is a key pathological feature. Tumors comprise multiple genetically-distinct cell populations, called clones. And the mutations of each clone are often of clinical significance. For instance, some co-mutations may give a clone an advantage over other cells, and thus enable its growth. Other mutational combinations may confer resistance to a given therapy. Thus, knowing the clonal composition of tumors is critical to understanding (and treating) cancer. Because bulk approaches do not measure cell-specific mutations, they cut out a major piece of the story. This begs the question of how relevant cancer models are if the clonal architecture remains unknown.
CRISPR and Single-cell DNA Sequencing Offer New Solutions
Fortunately, biotechnological advances are now enabling the development of more realistic cancer models. Nuclease-based gene-editing tools like CRISPR (short for: clustered regularly interspaced palindromic repeats) can change the genome in highly specific ways. For instance, CRISPR can be used to change nucleotides, add genetic sequences, or inactivate (knock out) genes. Moreover, these editors can be programmed to target multiple genes in the same cells in order to generate various co-mutations (multiplexing). This means researchers can generate genetic variation across cells, re-creating the heterogeneity of tumors.
Gene-editing technology has enabled the generation of more relevant cancer models. However, making a heterogeneous model is not very helpful unless one knows which cells contain which mutations. Although CRISPR can be used to target specific loci, mutational combinations are often generated randomly across cells. Thus, in order for researchers to understand the mutational architecture of the models they create, they must assess individual cells. This is where single-cell DNA sequencing (scDNA-seq) comes into play. Single-cell analysis platforms like Mission Bio’s Tapestri can be used to directly analyze the on-target (and predicted off-target) edits made to individual cells.1 This means that the CRISPR-generated co-mutations can be identified in each cell. Also, if engineered tumors are allowed to grow, scDNA-seq can be used to track the order of subsequent mutations and identify which clones expand over time.
New Study Models Acute Erythroid Leukemia (AEL)
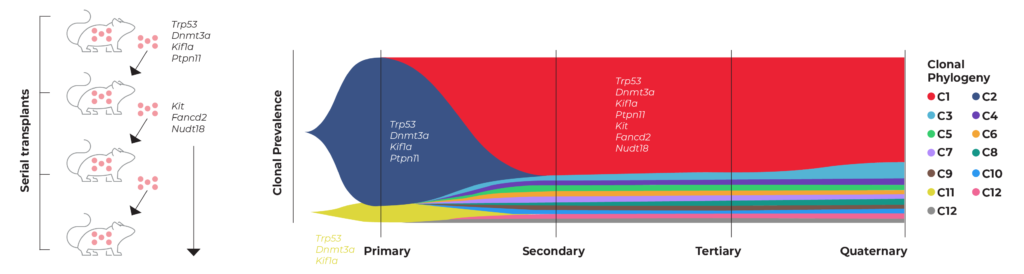
Dr. Ilaria Iacobucci, M.D. and her colleagues at St. Jude Children’s Research Hospital leveraged genome engineering and single-cell analysis to model cancer in a recent study published in Blood. Dr. Iacobucci’s team used CRISPR to generate multiple mutations typical of acute erythroid leukemia (AEL) in mouse hematopoietic stem and progenitor cells.2 The team then serially transplanted the edited cells into mice to evaluate how the tumors evolved over time. They used scDNA-seq to identify the primary (CRISPR-generated) and subsequent secondary mutations that drove AEL tumorigenesis in each transplant. Her team also evaluated an accompanying erythroid phenotype and used the models to assess drug sensitivity.
Interested in hearing more from the author herself? Join us for our webinar with Dr. Iacobucci on March 24th, 1 pm ET. Sign up here.
The work of Dr. Iacobucci on AEL modeling is also highlighted in our application note. As illustrated in the figure below, Iacobucci’s team tracked the clonal evolution of primary tumors (engineered using CRISPR) that were then serially transplanted into secondary, tertiary, and quaternary mice. From this study, the team was able to identify which clones (and mutations) drove the progression of each tumor.
Dr. Iacobucci’s study is not alone. Other researchers have also paired genome engineering and scDNA-seq to model various cancers, including chronic lymphocytic leukemia (CLL) and bladder cancer.3-4 It is encouraging to see how two powerful technologies are leveraged to advance pre-clinical cancer modeling. This approach holds great potential for enhancing our understanding of cancer, as well as enabling new therapies to be developed.
Interested in learning more about single-cell technology and its use in cancer research? Browse our free ebook.

References
- App Note: Measuring the efficiency of CRISPR genome-editing systems using the Tapestri Platform and Tapestri Single-Cell DNA Custom Panels. Mission Bio.
- Iacobucci I et al. Modeling and targeting of erythroleukemia by hematopoietic genome editing. Blood. 2021 Jan 19:blood.2020009103.
- ten Hacken E et al. High throughput single-cell detection of multiplex CRISPR-edited gene modifications. Genome Biol. 2020;21(1):266. Published 2020 Oct 20.
- App Note: Tapestri Platform Resolves Clonality of Heterogeneous Mouse Organoid Cancer Model through Single-Cell DNA Sequencing of Lentiviral Barcodes. Mission Bio.


