
Mission Bio is committed to helping you learn about single-cell DNA sequencing and multi-omics and how these exciting technologies are advancing medical science. Enjoy exploring our education hub!
CANCER & SINGLE-CELL ANALYSIS
Clonal Evolution in Cancer
Similar to Darwinian evolution at the species level, cancer is subject to natural selection and other evolutionary phenomena. Because genetic instability is a common feature of cancer cells, mutations are commonplace. Selection acts on this genetic variation, enabling cells with favorable phenotypes to propagate. Through an iterative process of mutation and cell expansion, tumors generate genetic and phenotypic heterogeneity. This cell-to-cell variation contributes to the complexity of the disease. In this section, we explore how tumors emerge and change over time.
Clonal Evolution Drives Tumor Heterogeneity
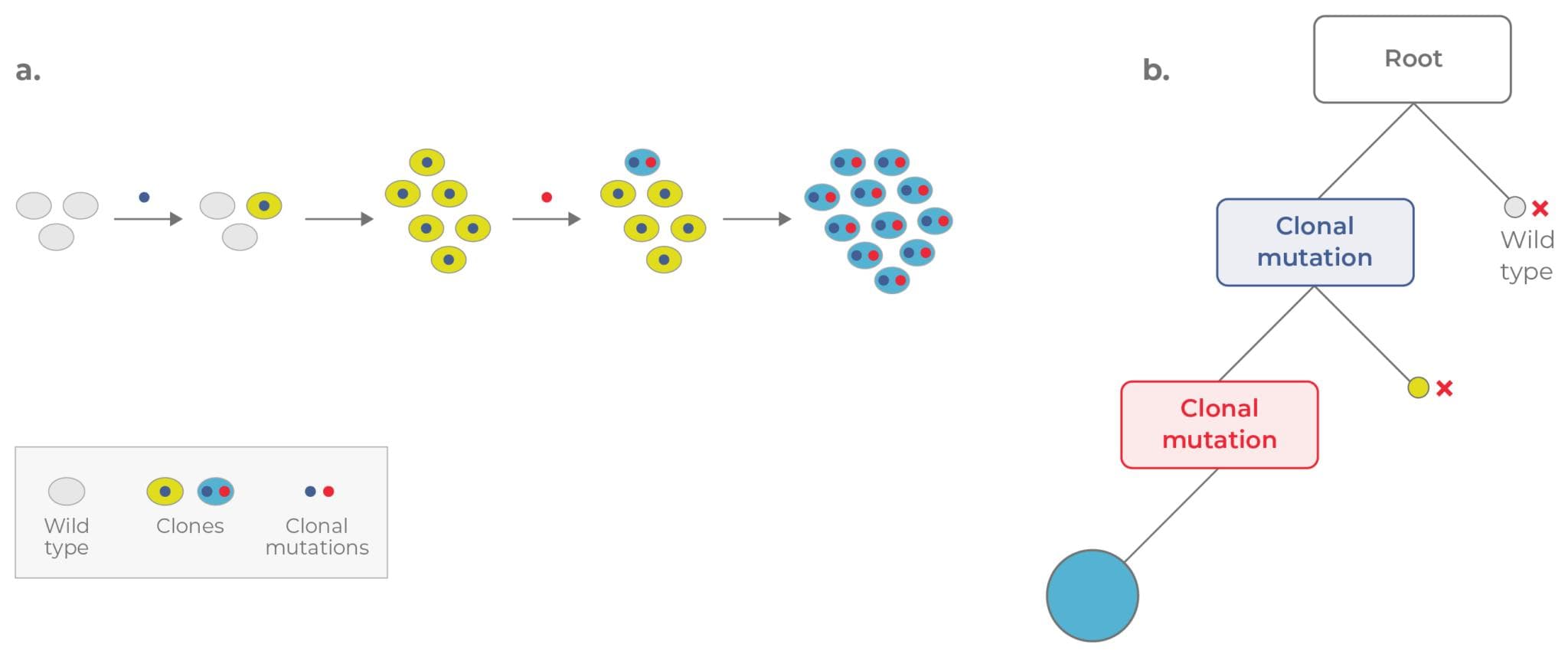
The path to cancer is often a long process with mutations accruing over years before a cell becomes malignant. Once the “right” combination of mutations initiates neoplastic change, however, a tumor begins to grow. There are two main models of clonal evolution in cancer: linear and branched.1 In linear evolution, each new driver mutation enables a selective sweep, in which the mutant cell — called a clone —outcompetes cells that don’t have the mutation. Mutations are accumulated in a cumulative and stepwise fashion as each new clone replaces the one before it (Fig 1). Note that in this model of clonal evolution, the cell population is largely homogeneous, with most cells containing the same clonal mutation(s) (i.e., passed down from a common parental clone).
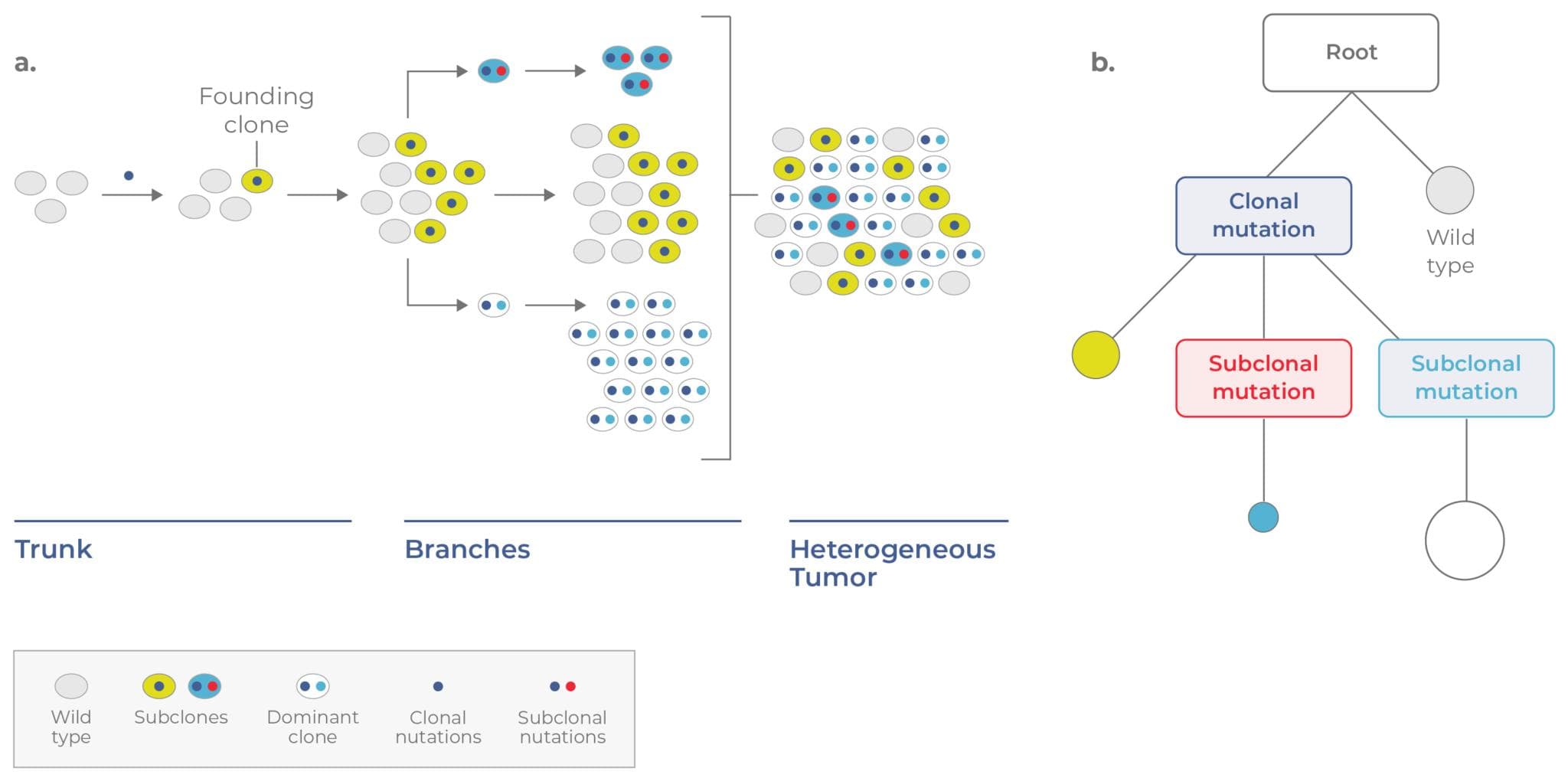
In contrast, tumors may undergo branched evolution, a process in which different cells independently acquire mutations (called subclonal mutations) that confer a fitness advantage. In this case, the population does not turn over with each new clone. Rather, multiple subclones co-exist and evolve in parallel. This process generates intratumoral diversity over time. To get a better idea of how this occurs, let’s walk through an example.
Consider a single healthy cell that acquires a driver mutation (e.g., IDH1) that enables its expansion. This cell is called a founding clone because it initiates tumorigenesis. As the clone propagates it gives rise to identical cells that contain the same clonal mutation. On a tumor phylogenetic tree, the founding clone resides on the “trunk” (Fig 2).
At a subsequent time point, a cell in the founding population may acquire a subclonal mutation, giving rise to a new subclone that propagates in parallel. Over time, more subclones may branch off, increasing the heterogeneity of the tumor. These genetically distinct subpopulations are represented by the “branches” on the tumor phylogenetic tree. In the example portrayed in Figure 2, three subclones co-exist together, with one dominant clone comprising the largest fraction of the tumor.
The evolutionary journey of each tumor is unique, with multiple factors determining the pattern of evolution (linear vs branched) and level of heterogeneity at any given point in time. Natural selection continues to act on the cells in the context of their microenvironment. Clonal populations with favorable phenotypes will increase in number, while those with neutral or deleterious characteristics remain stable or die out altogether.
Genetic drift and environmental changes also impact tumor composition. The introduction of an anti-cancer treatment into the tumor environment, for example, can dramatically remodel tumors by killing some cells and enabling the growth of others. Overall, the dynamic clonal evolution of tumors leads to their ever-changing clonal architecture.
Tumors Have Diverse Mutation Types
Clonal evolution in cancer may not only involve different genetic loci, but also different types of mutations. Variability in these mutations across cells also contributes to tumor heterogeneity. Tumor cells may contain a variety of lesions, ranging from small-scale mutations, like single-nucleotide variants (SNVs) and insertions/ deletions (indels) to large-scale chromosomal aberrations like translocations and gene fusions (Fig 3).
Although common in healthy cells, copy number variants (CNVs) occur more frequently in cancer. Types of CNVs include deletions, duplications, as well as loss of heterozygosity (LOH) (i.e. only one copy of an allele is present). Altering the number of gene copies — whether duplicating an oncogene or deleting a tumor suppressor — can result in uncontrolled cell proliferation.
As one might expect, cell-to-cell variation in mutation type and CNV increases the diversity of clones. This information may be clinically relevant, as the copy number of a particular gene may inform how virulent or drug-resistant a particular subclone is. For instance, the amplification of the oncogene CKS1B in multiple myeloma (MM) patients is associated with a poorer response to the chemotherapy, cisplatin.2 Knowing how genetic lesions vary across tumor cells thus gives a rare view into the strengths and weaknesses of tumors.

Clonal Evolution in Cancer Has Clinical Implications
Many studies to date indicate that clonal evolution in cancer is complex and leads to diverse clonal architecture. For instance, various whole-genome studies suggest that myeloid malignancies including acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), multiple myeloma (MM), myelodysplastic syndrome (MDS), and chronic lymphocytic leukemia (CLL) have a branched pattern of clonal evolution, in which multiple distinct subclones co-exist.3
This complexity makes cancer a formidable foe. Studies have shown that higher clonal diversity is associated with greater disease severity and poorer survivorship.4 Moreover, this variation often exists not only within a tumor, but also across tumors within a patient, and across patients in a population.
But untangling the complexity of cancer can even the playing field. Advances in single-cell technology are enabling researchers to precisely reconstruct tumor phylogenies, enabling the identification of the main mutational drivers of disease.4,5 Tumor heterogeneity also has profound implications for diagnosis, prognosis, and treatment of cancer. But before we jump into these applications, let’s first review how single-cell DNA sequencing and multi-omics are conducted.

Get a Free Quote
Speak to an expert today about the benefits of single-cell DNA and multi-omics analysis.
Vocabulary
Driver mutation: a mutation that is causally implicated in oncogenesis (gives the cell a growth advantage).
Clone: cells that are genetically identical.
Founding clone: clone that acquires the tumor-initiating mutation.
Subclone: a clone that is descended from another clone but has acquired additional mutation(s).
Dominant clone: the clonal population that occurs at the highest frequency in the tumor.
Clonal architecture: the subclones that comprise a tumor and their frequencies.
Clonal evolution: the process by which different subclones within a tumor change over time. Clonal evolution in cancer is shaped by stochastic factors and natural selection.
Single nucleotide variants (SNVs): variants characterized by a substitution of one nucleotide at a specific genomic position.
Insertions/ deletions (indels): insertion or deletion of nucleotides into the genome.
Copy number variants (CNVs): variants characterized by the number of repeated sections of the genome (deletions/duplications, loss of heterozygosity).
Loss of Heterozygosity (LOH): the loss of an allele from a heterozygous state, leaving only one allelic copy.
Translocation: a piece of one chromosome breaks off and attaches to another.
Gene fusion: a translocation event that places two formerly separate genes adjacent to one another. Some fusion genes encode fusion proteins that are implicated in cancer.