ENGINEERED CELLS & SINGLE-CELL ANALYSIS
Single-cell Analysis of Genome Engineering
Many of today’s cutting-edge research methods and therapies rely on the engineering of cells. By precisely altering DNA, genome engineering makes it possible to probe the relationship between a gene and its function, repair dysfunctional genes, or add functionality to cells. Changing the genome often involves some degree of variability. In some cases, different alterations are intentionally made across cells. Unwanted variation, on the other hand, may impact experimental results or therapeutic development. In this section, we discuss how single-cell DNA sequencing is used to measure precise genomic changes in individual cells.
Methods of Genome Engineering
Several genome engineering techniques can add exogenous genes or edit existing ones. In the former case, the addition of a transgene may offer new functionality or provide a working copy of a dysfunctional gene. Viral vectors, including adeno-associated viruses (AAV) and retroviruses (e.g., lentiviruses), are often used to ferry transgenes into cells and insert them into the genome (Fig 16a). DNA can also be edited directly using viruses or nuclease-based tools, such as CRISPR/Cas9 (Fig 16b). In this system, a single guide RNA (sgRNA) specifies a site in the genome for Cas9 to cut the DNA. Depending on the experimental goals, either homology-directed repair (HDR) or non-homologous end joining (NHEJ) is co-opted to make the desired edit while repairing the break. HDR is used to either change nucleotides or “knock in” new sequences, while NHEJ is used to disrupt genes (knockouts) by introducing indels into target sites. This latter method is useful for functional studies and loss-of-function (LOF) screens.
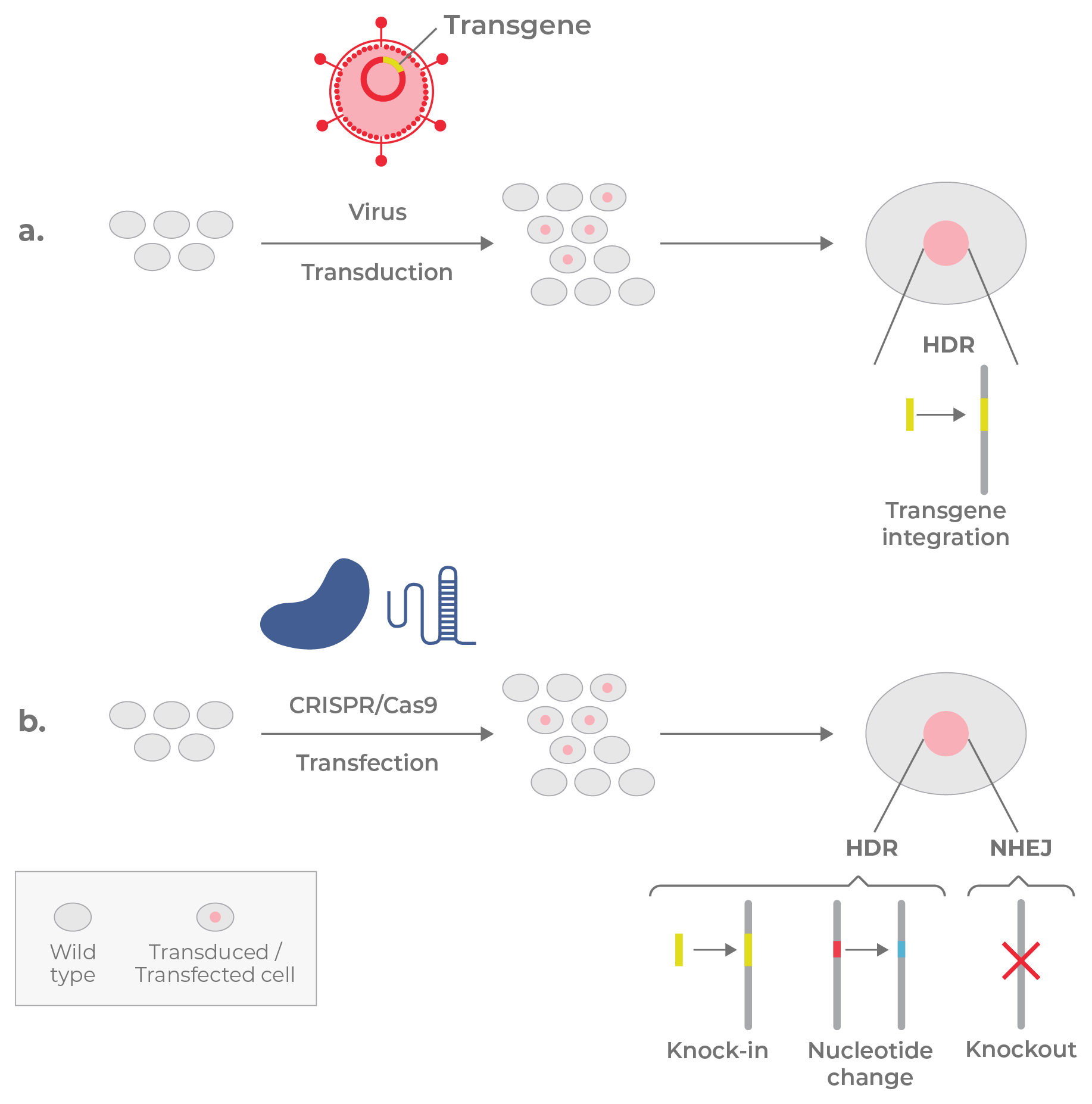
Figure 16. Genomic manipulation by viruses and gene editors. a. Viruses are often used to insert transgenes into the genomes of cells through HDR b. CRISPR/Cas9 is used to either insert sequences or change nucleotides through HDR disrupt gene function (knockout) through NHEJ.
Loss-of-function Screening
CRISPR loss-of-function (LOF) screens are an invaluable tool for functional genomics studies. These screens pair genome engineering (knockout of a large number of genes) with phenotypic analysis in order to characterize the genetic underpinnings of cellular pathways. LOF studies are crucial for understanding disease processes, discovering new drug targets, and assessing the genes that underlie the resistance or sensitivity of cancer to therapies.
Although the applications for CRISPR screening are quickly growing, typical screening protocols are limited in their ability to capture the heterogeneity of editing across cells. Typically, the average on-target editing efficiency is only measured at the population level before a screen. Thus, the effect of cell-to-cell variation for on- and off-target editing on phenotypic outputs is largely unknown.
Recently, there have been some advances in single-cell pooled screening with a transcriptome readout.20-23 While these screens have gained valuable insights, they also have some drawbacks. Significantly, the CRISPR-induced edits to the genome are not directly measured. Rather, the identity of the target is indicated by sequencing the integrated sgRNA sequence in cells. Thus, the type of edit is never identified, nor are any off-target changes to the genome.
The integration of single-cell DNA sequencing into screening workflows could solve this issue. Because single-cell DNA sequencing measures the actual changes to DNA, the on- and off-target edits can be directly identified and correlated with other analytes. With Tapestri technology, multi-omic screens that simultaneously measure genomic edits and cell-surface markers in individual cells are now possible. This enables the high-throughput annotation of mutations in different cell states associated with cancer.
Single-cell multi-omic screens that measure the genetic edits, transcriptome, and proteome within a single cell, are being developed. These advances in genome engineering and single-cell analysis will undoubtedly enhance our understanding of disease states and provide invaluable information for pharmaceutical development.
Disease Modeling
Disease modeling often involves recapitulating the underlying mutations of a disease in cells or organisms in order to study pathogenesis and test therapies. Different types of cancer are associated with different mutations in different genes, with clonal architecture adding an additional layer of complexity. Modeling cancer using cell or animal models means that these various levels of variation must be re-created. Fortunately, genome engineering tools can be used to generate mutations recurrent in different types of cancer. Furthermore, multiple mutations can be made to single cells in order to recreate co-mutated clones found in tumors.
Mutation combinations are often engineered in a random fashion across cells. Thus, genetic alterations must be analyzed in each cell in order to fully characterize the clonal composition of the model. Single-cell DNA sequencing can be used to directly analyze the genetic alterations produced by viruses and gene editors with single-cell resolution.24 The use of single-cell DNA sequencing has already been successfully used to evaluate several cancer models. In one example, Dr. John Lee (Fred Hutchinson Cancer Center) used lentivirus to generate random combinations of loss- and gain-of-function mutations across urothelial cells, building an organoid model of bladder cancer.25 After transplantation into mice and a period of in vivo selection, cDNA-seq was used to identify specific combinations of mutations that were highly correlated with tumorigenesis.
In another experiment, multiple mutations typical of acute erythroid leukemia (AEL) were generated in mouse hematopoietic stem and progenitor cells using genome engineering (CRISPR).26,27 After cells from a primary tumor were serially transplanted into mice, single-cell DNA sequencing was used to identify primary (CRISPR-generated) and secondary mutations that drove AEL tumorigenesis.
Dr. Ten Hacken et al. (2020) also used CRISPR to build a cancer model. Using a murine cell line, she generated LOF mutations recurrent in chronic lymphocytic leukemia (CLL) in random combinations.13 With single-cell DNA sequencing, her team was able to simultaneously measure zygosity, on-target, and off-target editing in single-target cells, as well as various patterns of co-mutation in multiplexed cells. Lastly, she compared changes in clonal architecture before and after edited cells were transplanted into mice. This study demonstrated the capability of single-cell DNA sequencing to analyze up to six concurrent targets in single cells, as well as predicted off-target activity.
Cell and Gene Therapies
Cell and gene therapies (CGTs) are rapidly expanding the treatment landscape for genetic diseases, including cancers and inherited diseases like sickle-cell anemia and Duchenne’s muscular dystrophy. Cell therapies involve treating disease by transferring cells with a desired function into the body. The cells may originate from the patient (autologous), a donor (allogeneic), or an established cell line. This approach requires that cells be collected outside the body (ex vivo) prior to introduction into the patient. Gene therapies involve the transfer of DNA or the direct alteration of the patient’s genome in order to treat a disease. Some therapies are classified as either a cell or a gene therapy, while others are a combination of both. For example, chimeric antigen receptor T-cell (CAR T-cell) therapy involves harvesting T-cells from a patient and genetically modifying them to express synthetic antigen receptors (CARs) on their surface. These receptors allow the T-cells to better bind to tumor cells so that they can kill them. The edited T-cells are re-infused into the patient, where they efficiently attack cancer cells.
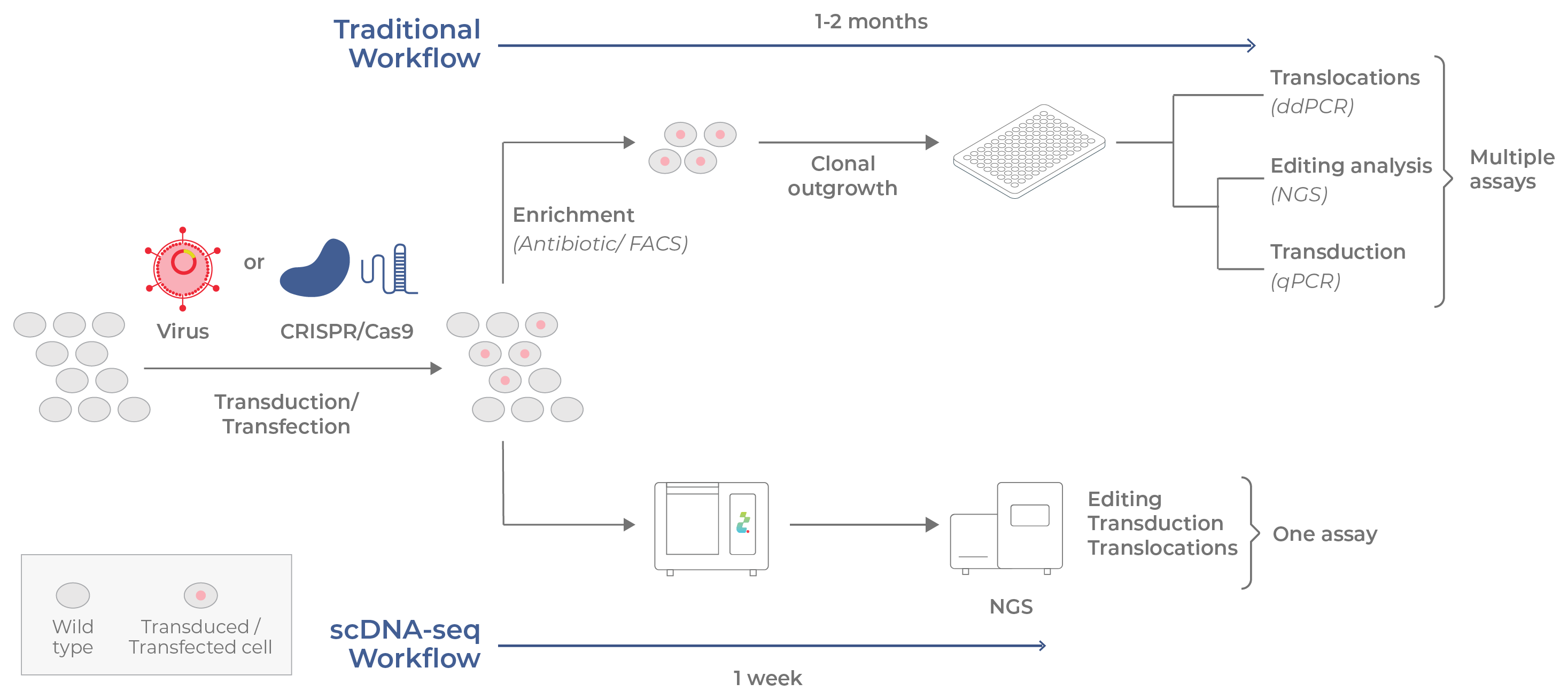
Figure 17. Conventional vs single-cell analysis on Tapestri used for cell and gene therapies. Cells of CGTs are assessed for variation in transduction efficiency, on- and off-target editing, and translocation events. Top: In the conventional workflow, transduced /transfected cells are enriched and clonally outgrown for several weeks. Assays that assess translocations and genome alterations are conducted separately. Bottom: With Tapestri, all analyses are combined in a single assay that can be completed in 1 week.
Viruses and gene editors are commonly used to modify the genomes of cells used in cell and gene therapies. Yet, the genetic heterogeneity produced by these alterations may have consequences to the efficacy and safety of the therapy. Analyzing single cells using conventional techniques is a long and laborious process (Fig 17, top). Genome engineering manipulations that involve viral or plasmid vectors often involve a time-consuming step to enrich successfully transduced/transfected cells. Furthermore, to identify cell-specific edits, individual cells must be separated and clonally outgrown (often taking weeks to complete). Once clones have been expanded, the analysis of genome alterations/ transduction efficiency is typically accomplished through sequencing or qPCR. Unwanted translocation events, which can potentially cause safety issues, require a separate assay (ddPCR or qPCR).
Single-cell DNA sequencing offers several advantages over traditional workflows (Fig 17, bottom). Because each cell is measured individually, the enrichment and clonal outgrowth steps can be bypassed, thus shortening the time between genome engineering and analysis from weeks to days. Additionally, the analysis of transduction efficiency/ editing and unintended translocation events can be combined into a single assay.28, 29
Single-cell analysis can be leveraged in multiple stages of the CGT pipeline, including protocol optimization, manufacturing, and release testing. By expediting these key steps, single-cell technology is helping advance the next generation of medicine.
Interested in Single Cell for Cell and Gene Therapy?
Capitalize on the power of single cell to characterize your cell and gene therapy products.
Learn MoreVocabulary
CRISPR/Cas9: a genome engineering tool composed of single guide RNA (sgRNA) and a Cas9 nuclease.
Homology-directed repair (HDR): a type of DNA repair that utilizes a DNA template to precisely repair double-strand breaks. It is leveraged in genome engineering to knock in nucleotides or make changes to the genome.
Non-homologous end joining (NHEJ): an error-prone type DNA repair that often inserts/deletes nucleotides at the break site. It is used in genome engineering to knock out genes.
CRISPR loss-of-function (LOF) screen: a screen that utilizes CRISPR to systematically ablate a large set of genes so that the phenotypic consequences can be assessed.
Disease modeling: the recapitulation of diseases using cell-based or organism-based models. This is often accomplished through genetic disruption or modification.
Cell and gene therapies (CGTs): a class of therapies that encompasses cell therapies (cells are used to introduce a therapy into the body), gene therapies (genetic alterations that have a therapeutic effect are made directly in the patient), or a combination of both.
Autologous: cells are sourced from the patient.







