
Mission Bio is committed to helping you learn about single-cell DNA sequencing and multi-omics and how these exciting technologies are advancing medical science. Enjoy exploring our education hub!
SINGLE-CELL APPLICATIONS | SPECIAL TOPICS
Beyond Bulk Sequencing: Uncovering Clonal Heterogeneity with Single-Cell Multiomic Cancer Profiling
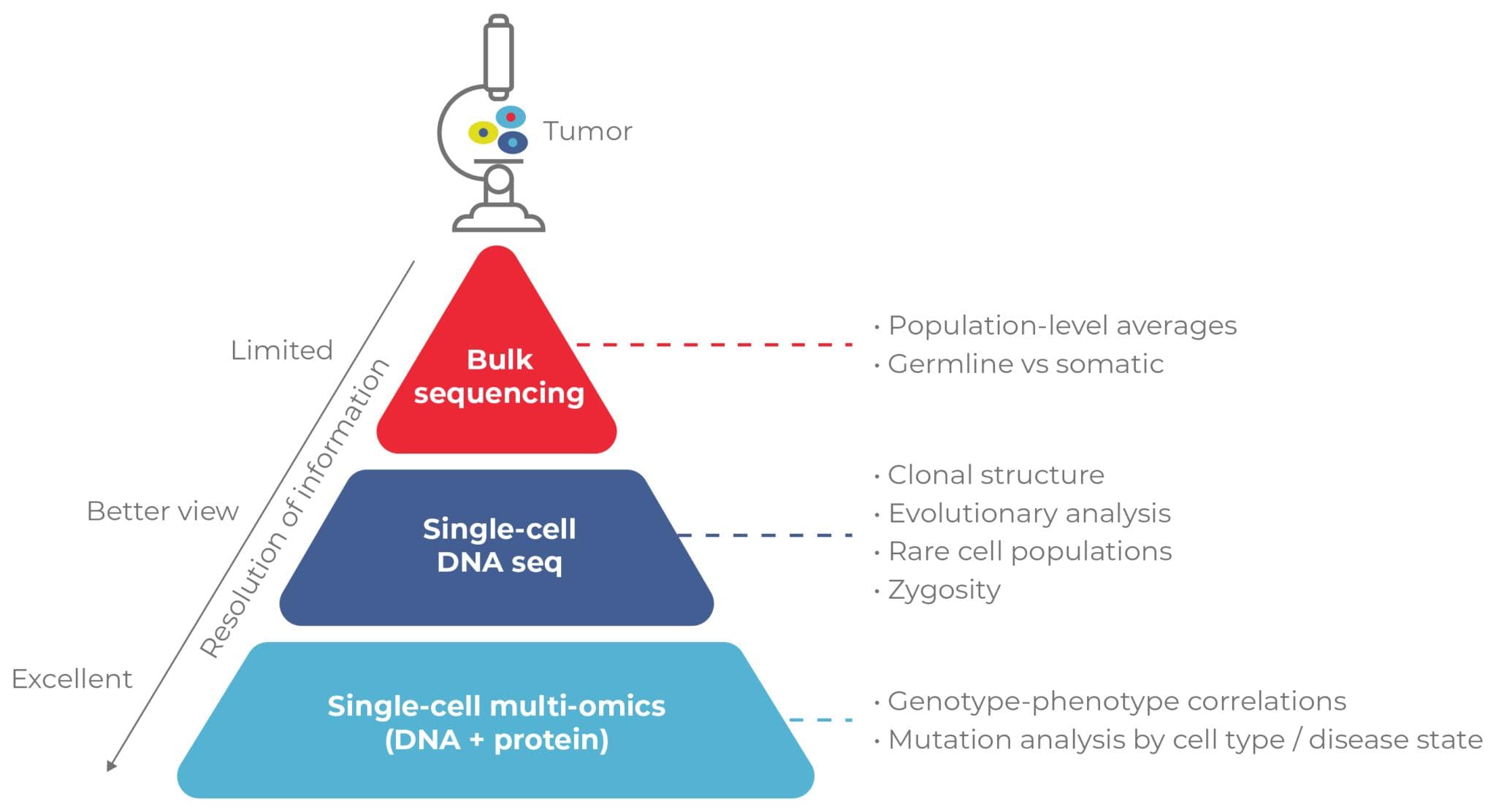
Though versatile, bulk sequencing provides a blurred view of cancer’s genetic landscape, obscuring the details that could make or break research directions and treatment strategies. Single-cell multiomic analysis brings those details into sharp focus, offering a powerful tool for understanding and combating clonal heterogeneity.
Here, we discuss the limitations of traditional cancer profiling methods and explore how a multiomic approach to single-cell analysis offers a powerful alternative. By providing a comprehensive overview of clonal architecture, this high-resolution method has profound implications for diagnosis, prognosis, and treatment strategy.
Genomic Instability Drives Clonal Heterogeneity
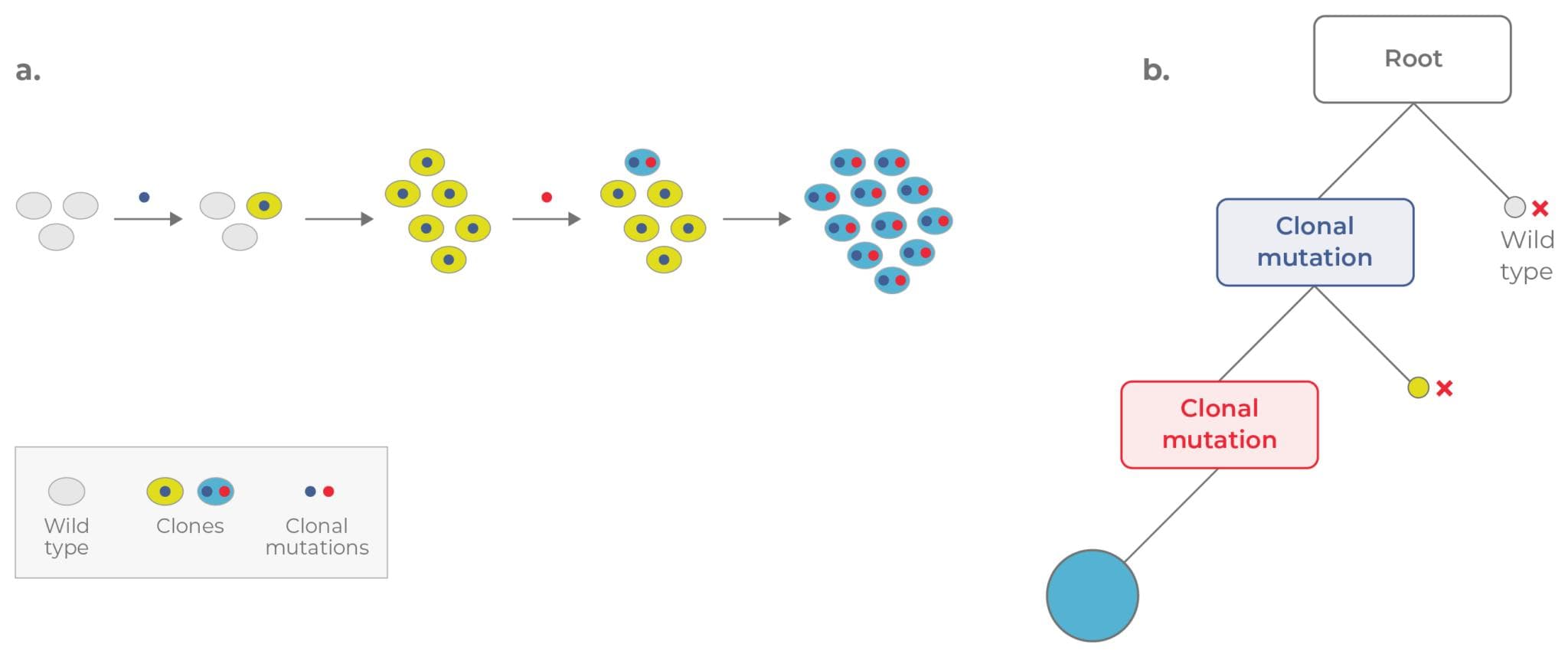
As cancer progresses, mutations accumulate–leading to subpopulations, or clones, with distinct genomes. This process, known as clonal evolution, allows subpopulations with selective advantages to outcompete others.
Clonal evolution underpins clonal heterogeneity (Fig. 1). Within a single patient, cancer cell populations can exhibit significant functional and phenotypic variation. Understanding this clonal heterogeneity is crucial for prognoses and treatment strategies. However, traditional methods are complex and costly.
Clonal Heterogeneity Informs Prognosis and Response
Higher clonal diversity is associated with greater disease severity, so recognizing and investigating these intrapatient differences is important for preclinical and clinical decision-making.
- Preclinical research: Understanding the clonal architecture (frequencies and mutations of subclones that comprise a tumor) supports the development of accurate and translational cancer models that improve predictions of treatment efficacy and resistance
- Clinical practice: Knowledge of clonal heterogeneity informs treatment strategies, allowing the deployment of more personalized and effective therapies
Clonal Heterogeneity in Solid Tumors
Genomic tumor profiling reveals clonal heterogeneity that can affect the growth and metastatic potential of tumors.1
For example, different subclones within single breast tumors may respond differently to hormone therapies, targeted treatments, or chemotherapy.2,3 Identifying these subclones will enable clinicians to prescribe combination therapies that successfully treat the entire tumor and adapt treatments as the tumor’s genetic landscape alters.
Clonal Heterogeneity in Blood Cancers
Clonal heterogeneity plays a significant role in disease progression, treatment response, and disease relapse in blood cancers.4,5
Some heme malignancies, such as multiple myeloma (MM), are especially genetically diverse, causing them to have high relapse rates.6 Understanding the complex clonal composition of a patient’s MM from diagnosis and beyond helps clinicians select the most effective treatment regimen and monitor for the emergence of resistant clones.
Traditional Cancer Profiling Methods Lack Resolution and Integration
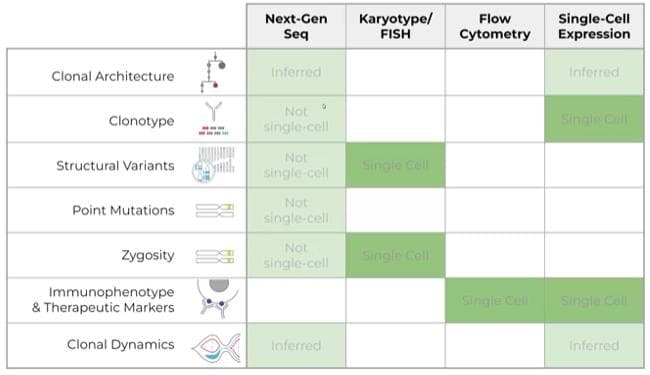
Standard methods for investigating clonal heterogeneity include bulk sequencing and flow cytometry (Fig. 2).7 Although these techniques have been instrumental in advancing the understanding of cancer genomics, they are often limited to focusing on isolated cellular attributes and unable to provide insight into clonal heterogeneity.
Bulk Sequencing: Versatile but Low-Resolution
Bulk sequencing, such as next-generation sequencing (NGS), averages genetic information across all cells in a sample, preventing the identification of subclones and their specific mutations. NGS is also susceptible to inaccurate profiling when dominant clones mask less frequent but potentially crucial subclonal mutations.
These limitations result in an incomplete understanding of cancer’s genetic diversity. This is associated with an increased likelihood of selecting unsuitable therapies that don’t target the entire cancer population, inadvertently promoting the growth of resistant clones.
Single-Cell Analyses: Focused but Fragmented
Many single-cell cancer profiling assays focus on specific aspects of cellular biology (e.g., immunophenotype, zygosity) and lack multiomic capabilities. Therefore, researchers must perform multiple assays. Not only is this costly, but it exhausts precious samples, leaving little material for downstream analyses.
This approach requires complex integration of data obtained from separate assays to achieve a global view of clonal heterogeneity. Harmonizing datasets from distinct assays typically requires sophisticated computational methods and bioinformatics expertise to ensure accurate alignment and reliable interpretation.
Single-Cell Multiomic Analysis Elevates Cancer Profiling with Unparalleled Detail
Comprehensive multiomic analysis of single cells addresses the challenges associated with traditional cancer profiling methods by offering a holistic view of clonal heterogeneity.
This approach provides a high-resolution and integrated understanding of cancer biology by simultaneously analyzing multiple molecular modalities—such as DNA, RNA, and proteins—within individual cells (Fig. 3).
Advantages of Single-Cell Multiomic Analysis
- High resolution: Identifying and characterizing individual subclones reveals their unique genetic, transcriptomic, and proteomic profiles. This provides detailed insights into their distinct vulnerabilities and behaviors, informing treatment strategy and prognosis.
- Accurate mutational profiling: Accurately capturing genetic mutations at the single-cell level provides a complete picture of the cancer’s genetic landscape. By detecting even rare subclonal mutations, treatment can be tailored to modulate multiple therapeutic targets if necessary.
- Comprehensive insights: Integrating data from multiple molecular layers enables researchers to identify mutually exclusive or co-occurring features in a single assay, enhancing their understanding of how different mutations and pathways interact.
Clinical Implications of Single-Cell Multiomic Analysis
- Target identification: By characterizing the genetic and molecular profiles of individual subclones, researchers can identify novel therapeutic targets. Insight into which targets are typically co-expressed or mutually exclusive fuels the development of more effective treatments.
- Treatment stratification: Understanding a patient’s clonal architecture allows for the design of personalized treatment regimens that address the unique vulnerabilities of each subclone.
- Monitoring response: Cancer profiling is not only a useful diagnostic tool but also essential for tracking patient response to treatment. Understanding this response at a subclonal level provides valuable information for adjusting treatment strategies to overcome resistance.
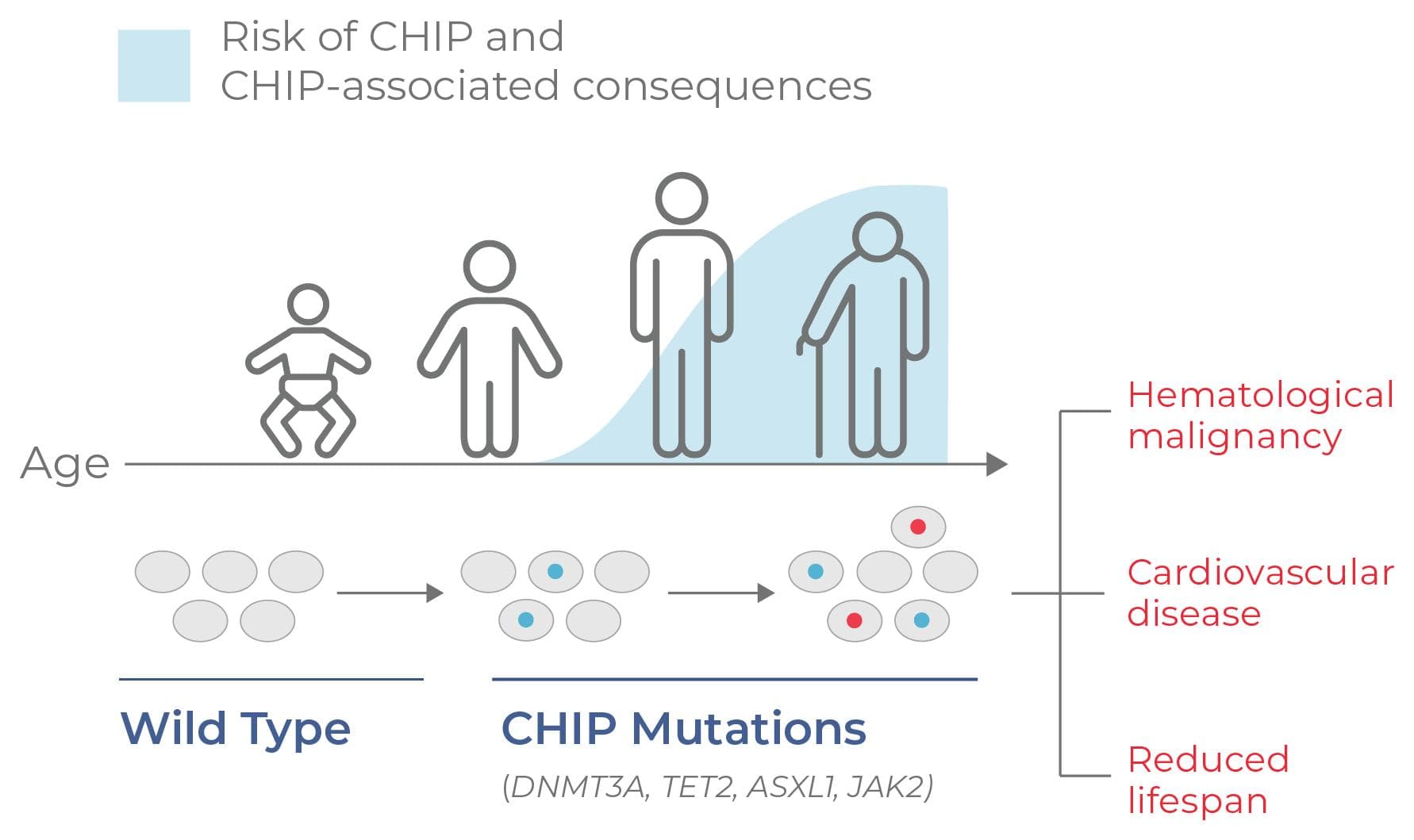
- Understanding risk: Multiomic analysis of single cells aids in understanding potential prognostic conditions, such as clonal hematopoiesis of indeterminate potential (CHIP) (Fig. 4). Clonal profiling in people with CHIP helps detect disease-driving mutations, improving predictive models and personalized treatment plans.
Given these multifaceted advantages, it is crucial to further demonstrate the clinical research utility of a multiomic approach to single-cell analysis.
Building the Evidence: Validating Single-Cell Multiomic Analysis in Clinical Research
Applying a multiomic approach at single-cell resolution can significantly enhance heme malignancy and genomic tumor profiling by providing detailed and integrated insights into complex cancer biology. Robust data and validation processes are essential to prove its utility and value as a clinical research tool.
Demonstrating the benefits of multiomics at the single-cell level in clinical research involves conducting retrospective and prospective studies to assess how this technology aids understanding of clonal architecture to enhance the prediction of disease progression, biomarker insights, and therapeutic target identification.
Comparative studies that showcase the increased sensitivity of multiomic analysis compared to current single-cell methods (e.g., flow cytometry for acute myeloid leukemia) will highlight the advantages of this approach in predicting disease progression, therapeutic response, and relapse.
Ultimately, continuous monitoring and documentation of the performance and outcomes of single-cell multiomic analysis in real-world translational and clinical research will further substantiate its benefits, paving the way for its widespread adoption.
Single-cell multiomic analysis stands at the forefront of cancer profiling, offering unparalleled insights into clonal heterogeneity and significantly enhancing our understanding of cancer biology. By overcoming the limitations of traditional methods, this advanced approach paves the way for more precise insights, personalized treatments, and improved patient outcomes.
Are you ready to join the resolution revolution?
DRIVE ACTIONABLE INSIGHTS ACROSS CANCER RESEARCH WITH SINGLE-CELL MULTIOMICS
For Research Use Only. Not for use in diagnostic procedures.
References
- Lüönd F, Tiede S, Christofori G. Breast cancer as an example of tumour heterogeneity and tumour cell plasticity during malignant progression. Br J Cancer. 2021;125(2):164-175. doi:10.1038/s41416-021-01328-7
- Rye IH, Trinh A, Sætersdal AB, et al. Intratumor heterogeneity defines treatment‐resistant HER2+ breast tumors. Mol Oncol. 2018;12(11):1838-1855. doi:10.1002/1878-0261.12375
- Marusyk A, Janiszewska M, Polyak K. Intratumor heterogeneity: the Rosetta stone of therapy resistance. Cancer Cell. 2020;37(4):471-484. doi:10.1016/j.ccell.2020.03.007
- Aertgeerts M, Demeyer S, Lamote J, et al. Clonal Heterogeneity and Evolution during Treatment in High Hyperdiploid B-Cell Acute Lymphoblastic Leukemia. Blood. 2023;142(Supplement 1):1606. doi:10.1182/blood-2023-180760
- Benard BA, Leak LB, Azizi A, Thomas D, Gentles AJ, Majeti R. Clonal architecture predicts clinical outcomes and drug sensitivity in acute myeloid leukemia. Nat Commun. 2021;12(1):7244. doi:10.1038/s41467-021-27472-5
- Bianchi G, Ghobrial IM. Biological and Clinical Implications of Clonal Heterogeneity and Clonal Evolution in Multiple Myeloma. Curr Cancer Ther Rev. 2014;10(2):70-79. doi:10.2174/157339471002141124121404
- Malone ER, Oliva M, Sabatini PJB, Stockley TL, Siu LL. Molecular profiling for precision cancer therapies. Genome Med. 2020;12:8. doi:10.1186/s13073-019-0703-1